
This is is the nanomerge pipeline from the Sequana project
| Overview: | merge fastq files generated by Nanopore run and generates raw data QC. |
|---|---|
| Input: | individual fastq files generated by nanopore demultiplexing |
| Output: | merged fastq files for each barcode (or unique sample) |
| Status: | production |
| Citation: | Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352 |
You can install the packages using pip:
pip install sequana_nanomerge --upgrade
An optional requirements is pycoQC, which can be install with conda/mamba using e.g.:
conda install pycoQC
you will also need graphviz installed.
sequana_nanomerge --help
If you data is barcoded, they are usually in sub-directories barcoded/barcodeXY so you will need to use a pattern (--input-pattern) such as */*.gz:
sequana_nanomerge --input-directory DATAPATH/barcoded --samplesheet samplesheet.csv
--summary summary.txt --input-pattern '*/*fastq.gz'
otherwise all fastq files are in DATAPATH/ so the input pattern can just be *.fastq.gz:
sequana_nanomerge --input-directory DATAPATH --samplesheet samplesheet.csv
--summary summary.txt --input-pattern '*fastq.gz'
The --summary is optional and takes as input the output of albacore/guppy demultiplexing. usually a file called sequencing_summary.txt
Note that the different between the two is the extra */ before the *.fastq.gz pattern since barcoded files are in individual subdirectories.
In both bases, the command creates a directory with the pipeline and configuration file. You will then need to execute the pipeline:
cd nanomerge sh nanomerge.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s nanomerge.rules -c config.yaml --cores 4 --stats stats.txt
Or use sequanix interface.
Concerning the sample sheet, whether your data is barcoded or not, it should be a CSV file
barcode,project,sample barcode01,main,A barcode02,main,B barcode03,main,C
For a non-barcoded run, you must provide a file where the barcode column can be set (empty):
barcode,project,sample ,main,A
or just removed:
project,sample main,A
With apptainer, initiate the working directory as follows:
sequana_nanomerge --use-apptainer
Images are downloaded in the working directory but you can store then in a directory globally (e.g.):
sequana_nanomerge --use-apptainer --apptainer-prefix ~/.sequana/apptainers
and then:
cd nanomerge sh nanomerge.sh
if you decide to use snakemake manually, do not forget to add apptainer options:
snakemake -s nanomerge.rules -c config.yaml --cores 4 --stats stats.txt --use-apptainer --apptainer-prefix ~/.sequana/apptainers --apptainer-args "-B /home:/home"
This pipelines requires the following executable(s), which is optional:
- pycoQC
- dot
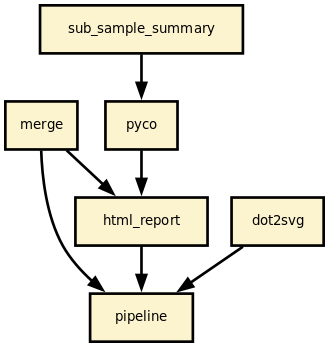
This pipeline runs nanomerge in parallel on the input fastq files (paired or not). A brief sequana summary report is also produced.
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
| Version | Description |
|---|---|
| 1.5.1 |
|
| 1.5.0 |
|
| 1.4.0 |
|
| 1.3.0 |
|
| 1.2.0 |
|
| 1.1.0 |
|
| 1.0.1 |
|
| 1.0.0 | Stable release ready for production |
| 0.0.1 | First release. |