4. Projects
A project includes a set of patients or cell line experiments. The project views provide the statistics for the whole cohort and help users identify important events with integrated user interface.
The summary tab shows the meta data statistics. Oncogenomics automatically determines if the data types is categorical or numeric and shows bar chart or pie chart respectively. If the project has diagnoses >= 10, a sortable table will be displayed.
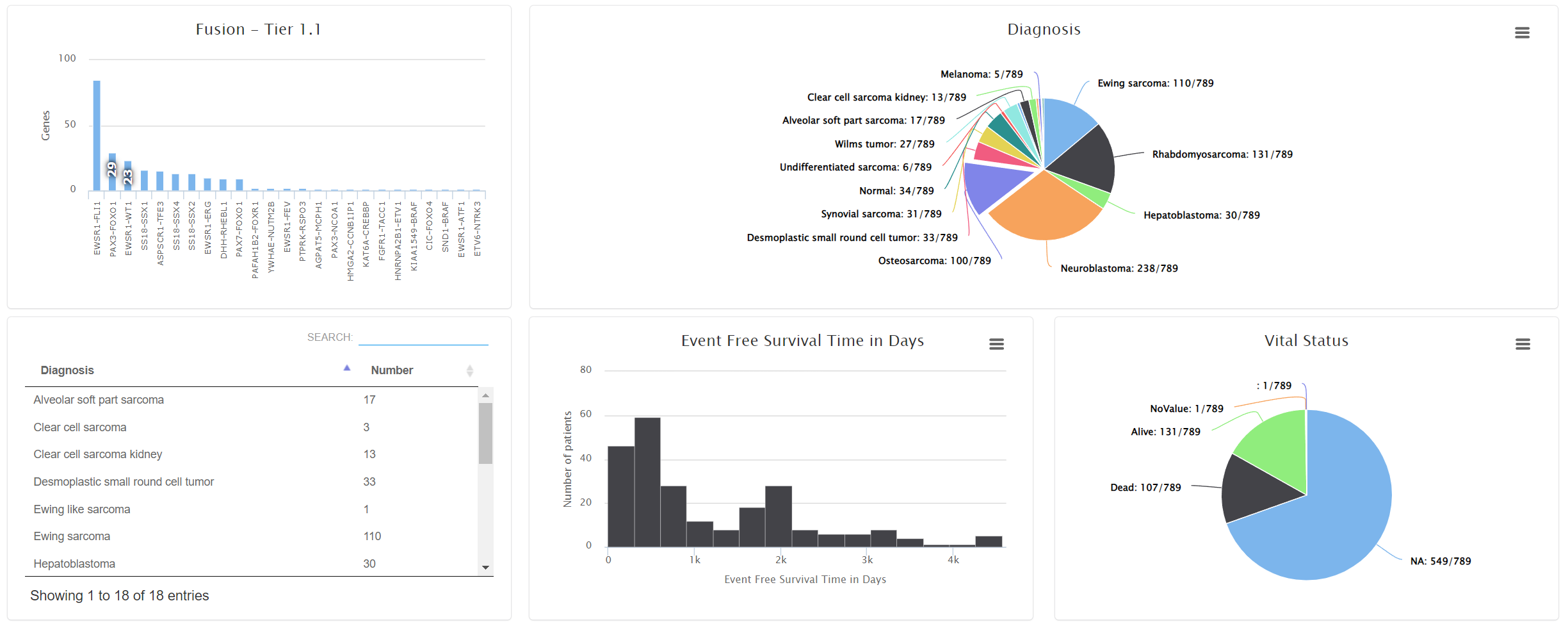
Users can move the mouse over the bar/pie area to see the detailed patient count. By clicking them, a patient table will show up. Users can view the patient pages by selecting the links.
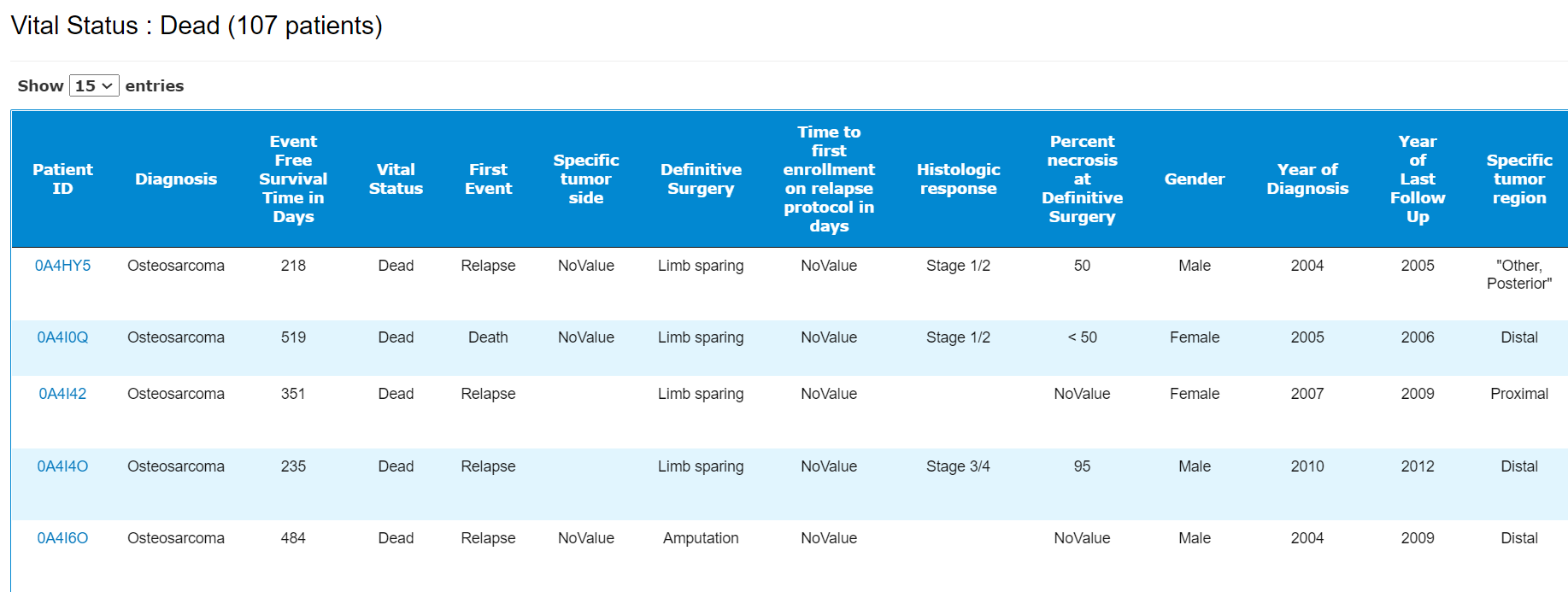
A patient table including all the meta data. By clicking , the sample list will be displayed.
The mutations tab shows the summary of the mutations called in the project categorized into the following types:
- Germline: for paired tumor/normal DNA-seq, the variants specific to normal sample.
- Somatic: for paired tumor/normal DNA-seq, the variants specific to tumor sample.
- Tumor: the variants for tumor only DNA-seq samples.
- RNAseq: the RNAseq variants.
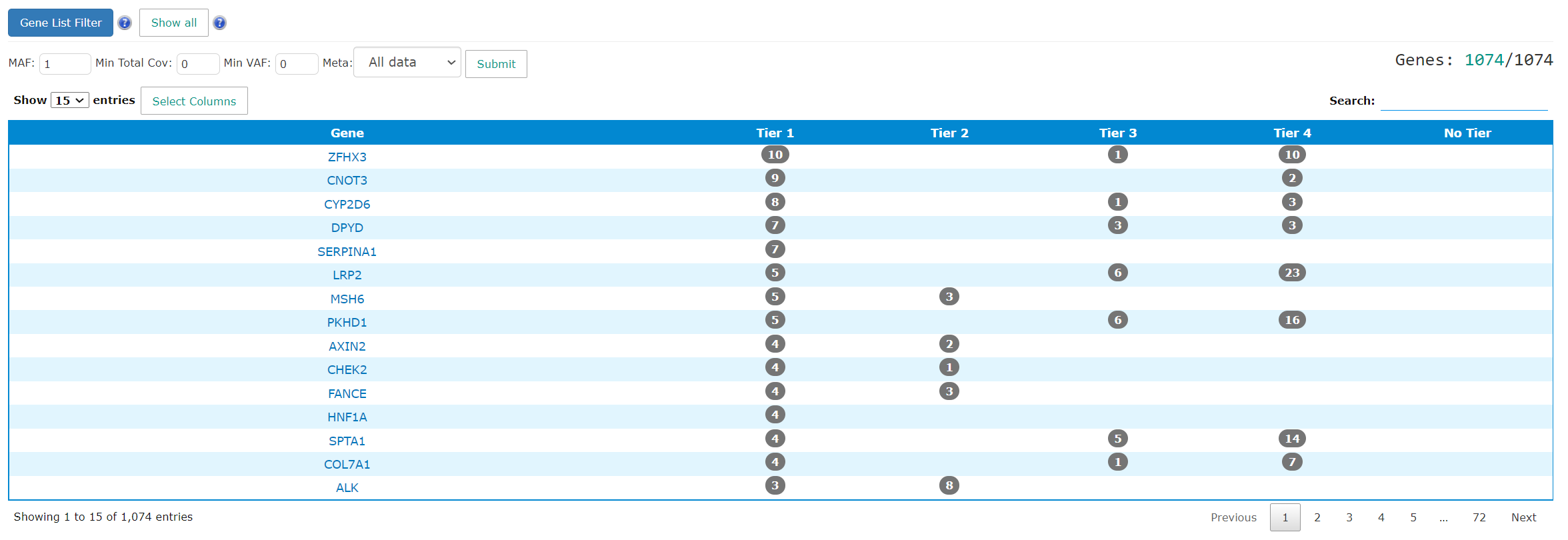
The summary table includes the gene name and the patient count for each tier. The gene name links will direct users to gene summary page. The number in the tiering columns refer to how many patients have mutations in the genes. The link takes users to the gene level mutation pages. The results can be filtered by MAF, coverage, VAF and meta data.
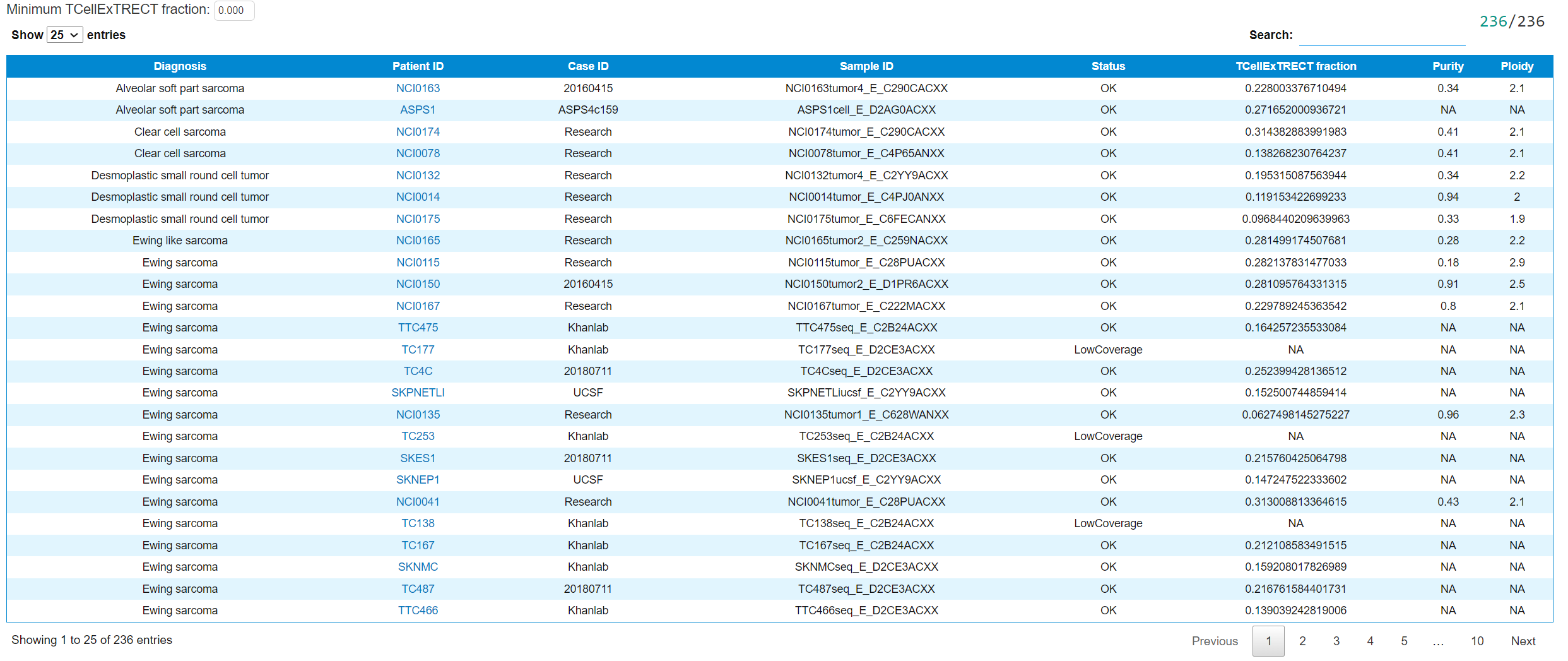
This tab shows the table summary of the T-cell infiltration using TcellExTRECT. The detailed explanation of the content can be found in case wiki page. The table can be filtered by the minimum T-cell fraction.
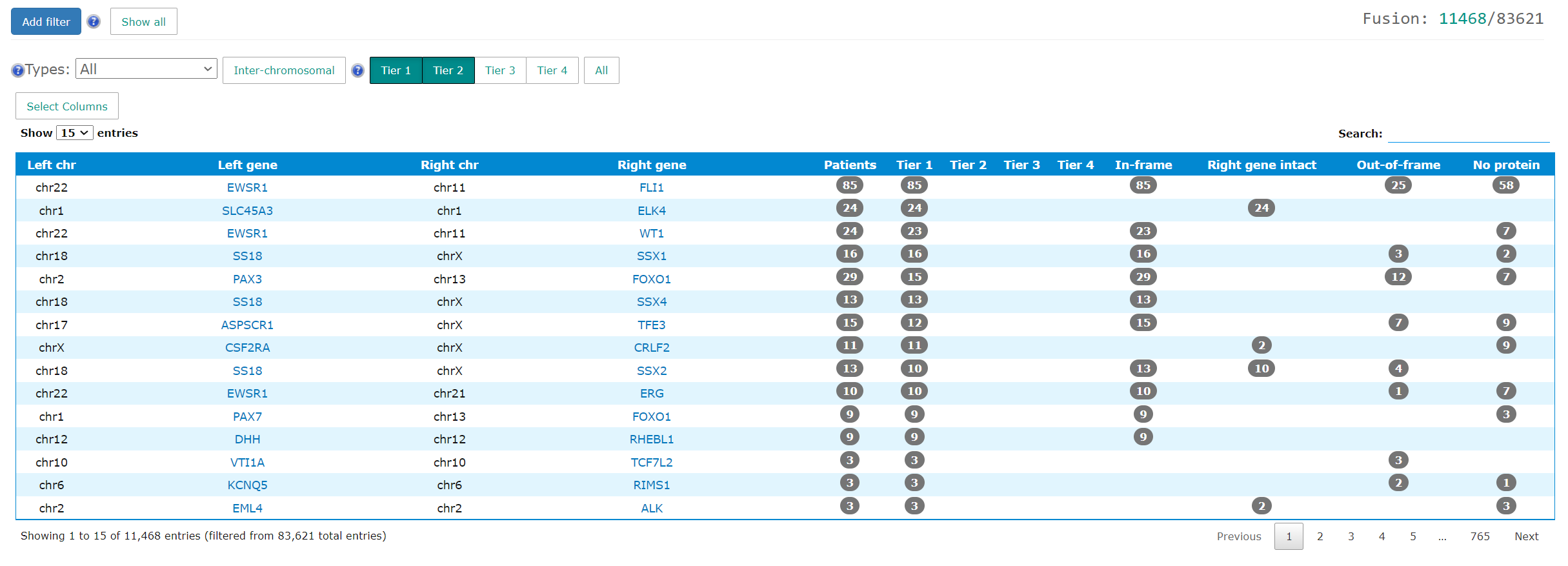
The project level fusion tab summarizes all fusions found in the project. The loading time may be a bit longer if the number of fusions is big. In addition to the left/right gene names and chromosomes, the table shows the patient count grouped by tiering and fusion types. The detailed information is available in case wiki page.
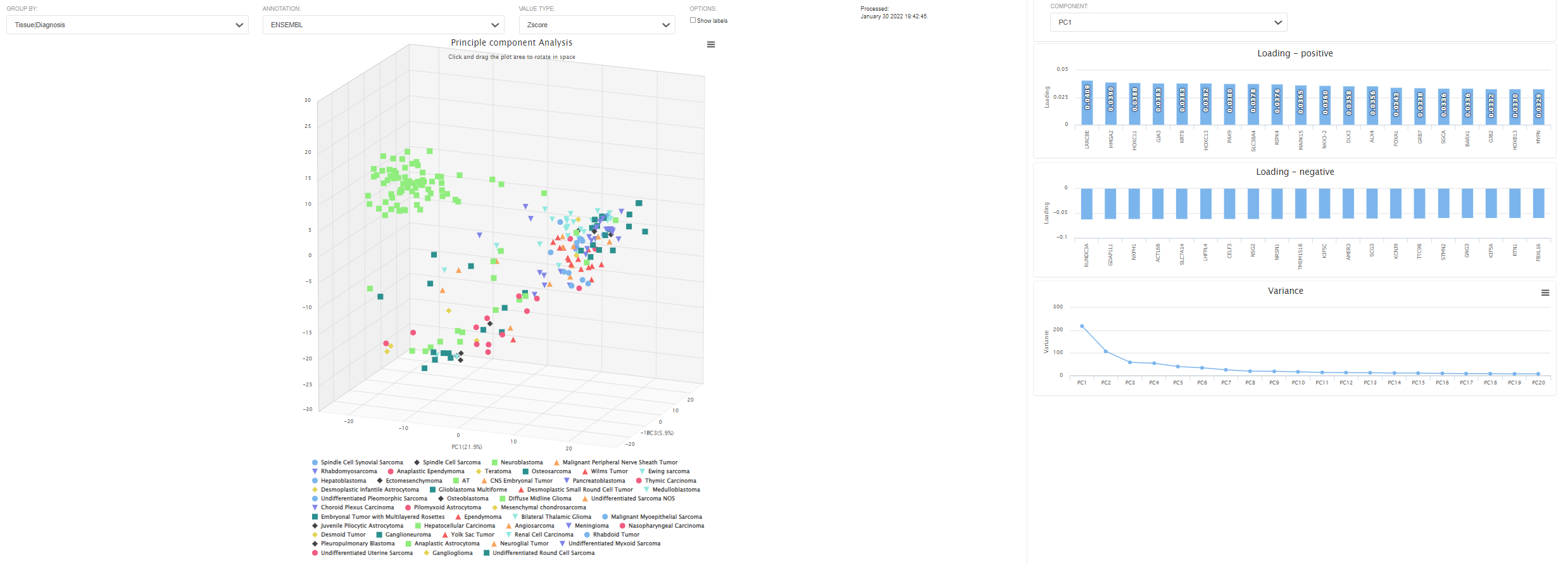
The project level expression tab shows the the principle component analysis and heatmap plot.
The pre-calculated PCA is based on 1000 genes with highest variants. The values are log2-CPM. This 3D plot can be colored by meta data. Users can also use z-scored or log2 values. Each dot represents an RNAseq sample. By clicking "show labels", the sample ID will be displayed. This might take a while if the number of samples is large. The loading and the total variance will also be shown on the side panel.
This tab shows the expression heatmap. User can switch the value types to TMM-normalized FPKM or TPM. The order can be sorted by either genes or samples by clicking them.
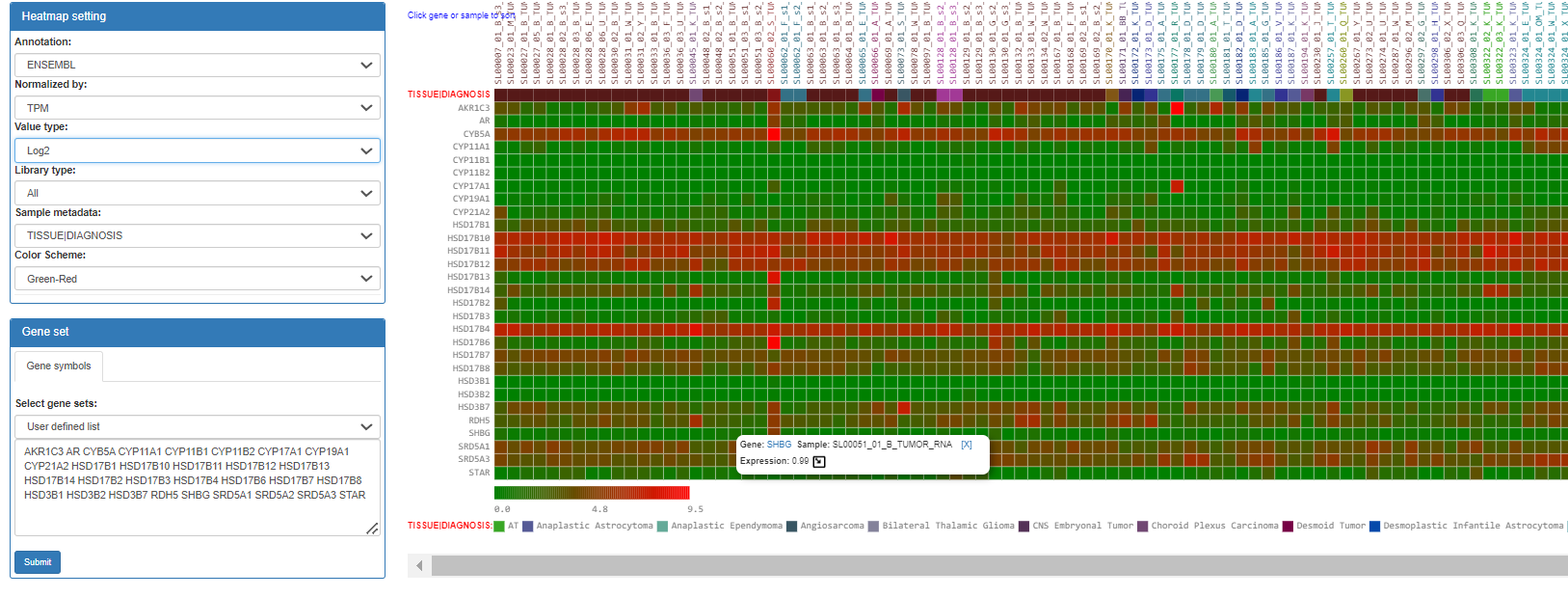
The detailed information will be shown as a popup window when the cursors move to the cells. When clicking 
Users can download expression matrices in this tab. This is particular useful when users need the raw count data to perform downstream analyses such as differential expression analysis.
In summary tab, Oncogenomics summarizes Sequenza results for each case. Each row represents a case. The definition of columns can be found in case wiki page.
In download tab, there are four types data can be downloaded:
- Sequenza summary: the summary information as in summary tab.
- Sequenza files: a zipped file of all the Sequenza output files in the project.
- Sequenza matrix file: Sequenza CNV file in matrix format. Rows are gene and columns are samples. The values are CN defined in Sequenza.
- CNVkit matrix file: CNVkit CNV file in matrix format. Rows are gene and columns are samples. The values are log ratio defined in CNVkit.